
The concept of in vitro Area Under the Concentration Time Curve (AUC) in drug discovery
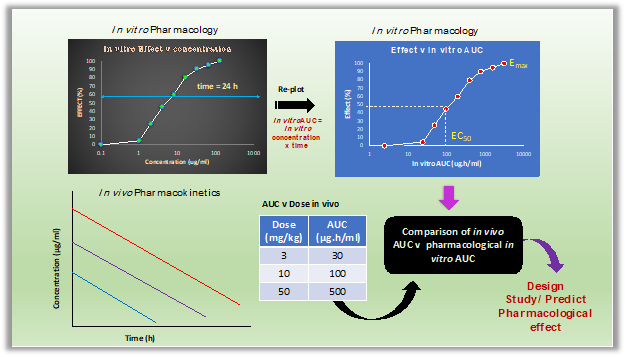
In drug discovery, in vitro pharmacology studies are fundamental in assessing a drug candidate’s intrinsic efficacy (Emax,activation or Imax,inhibition) and potency (EC50activation or IC50inhibition) on its pharmacological (drug) target outside the cells (binding and inhibiting or activating the target’s activity e.g. enzyme and receptor assays) and inside the cells (functional assays such as cell division, growth, inhibition, killing, differentiation, apoptosis). In a typical assay, the efficacy and potency of the drug candidate is evaluated by incubating the target or cells with different concentrations of the drug (6-8 concentrations) for a defined period of time (24-72 h), and measuring the desired effect (end point) such as enzyme or receptor activity and effects on cells expressing the target for functions such as cell proliferation. The efficacy (Emax or Imax) and potency (EC50 or IC50) is estimated by plotting the effect against concentration. Another type of assay measures the effect of the candidate drug over time where the pharmacological effect is measured at different time points against different concentrations. The aim of this assay is to evaluate the time course of pharmacological effect over time at different concentrations (eg. in vitro kill kinetics of bacterial and cancer cells).
In both the assays, the effect is evaluated against varying concentrations over a fixed time period. The concentrations associated with Emax or Imax and EC50 or IC50 are then used in combination with PK (Cmax, AUC, time of concentrations exceeding potency) to assess potential for efficacy in pharmacology models. Time is assumed to be constant, and therefore usually not considered as relevant for pharmacological activity. However, in reality, the pharmacological or toxicological effect of a drug is usually dependent both on its concentration and time for it is exposed to the cells containing the target.
It is important to note that, in in vivo studies both in animals and humans, AUC is an important PK parameter used to correlate PK with pharmacological and toxicological effects. For example, dose-exposure-response relationships are used in quantifying relationships between PK and PD (pharmacodynamics), i.e., PK/PD relationships. The AUCs for the no-adverse-effect dose level (derived from toxicology) and pharmacologically active dose level (from pharmacology studies) are used in estimating the therapeutic index or the safety margin. In infection, the pharmacological effect of drugs can be driven by exposure (AUC, Cmax) or by time for which unbound concentrations exceed the potency in a dosing interval.
Given the importance of AUC in in vivo studies, can it be estimated in vitro and used to assess the candidate’s potential to show efficacy/safety based on its PK (AUC) data?
The in vitro AUC is defined as the time for which a candidate drug’s concentration is exposed to the desired cells or tissues or organs in the in vitro assay. It is estimated as the product of the concentration and the time of exposure (In vitro AUC = Concentration x time) and has the same units as the in vivo AUC like µg.h/ml or ng.h/ml.
Thus, if there are 8 concentrations in the assay for which the effect is determined, there will be eight in vitro AUCs associated with effect. One can then estimate the in vitro AUC associated with Emax and EC50. Based in the in vitro AUC, it is possible to predict pharmacological exposures (AUC in vivo) required to achieve a specific magnitude of effect in vivo, assuming there is a correlation between in vitro and in vivo pharmacological effect. The estimation of in vitro AUC assumes that the concentration of candidate drug in the assay does not change significantly in the time period (inhibition or activation assays with cells expressing the target).
The concept of in vitro AUC was used in characterizing the relationships between in vitro, ex vivo and in vivo PK/PD of Rifampicin and Isoniazid on Mycobacterium tuberculosis.
References:
- Jayaram R, Gaonkar S, Kaur P, Suresh BL, Mahesh BN, Jayashree R, Nandi V, Bharat S, Shandil RK, Kantharaj E, Balasubramanian V. 2003. Pharmacokinetics-pharmacodynamics of rifampin in an aerosol infection model of tuberculosis. Antimicrob Agents Chemother. 47(7):2118-24.
- Jayaram R, Shandil RK, Gaonkar S, Kaur P, Suresh BL, Mahesh BN, Jayashree R, Nandi V, Bharath S, Kantharaj E, Balasubramanian V. 2004. Isoniazid pharmacokinetics-pharmacodynamics in an aerosol infection model of tuberculosis. Antimicrob Agents Chemother. 48(8):2951-7
Ramesh Jayaraman
DoseQuantics Consulting Pvt Ltd