
The Hollow Fiber Infection Model in Anti-Bacterial Drug Discovery and Development
Ramesh Jayaraman, DoseQuantics Consulting
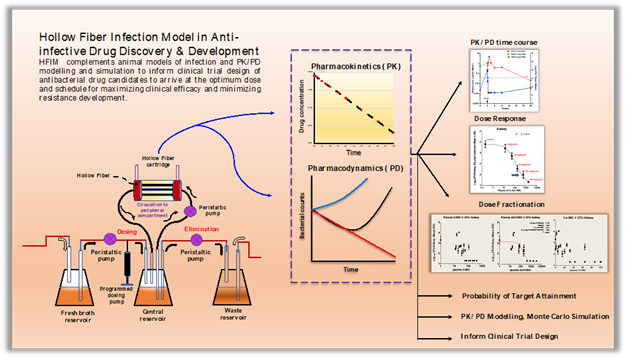
In anti-bacterial drug discovery and development, preclinical animal models of infection have played a pivotal role in evaluating the translational potential of drug candidates. Drug regulatory agencies review pharmacology data from preclinical animal models of infection for understanding the pharmacokinetic/pharmacodynamic (PK/PD) basis for setting efficacious dose and regimens for phase 2 and phase 3 clinical trials. Indeed, it has been shown that the PK/PD measures of efficacy in animal models are strongly predictive of clinical efficacy.
However, the difficulty to mimic human PK in preclinical animal models, lack of appropriate animal models of infection for specific pathogens, and animal ethics reasons, has led to the development of in vitro non-animal models for evaluating the PK/PD of anti-bacterial agents. The hollow fiber infection model (HFIM) is one such model which evaluates anti-bacterial effects under dynamic drug concentrations (PK).
The HFIM consists of a series of compartments that are inter-connected with peristaltic pumps which regulate the rate of inflow and outflow of liquid. The basis setup has a reservoir containing growth (nutrient) medium connected to a central vessel which in turn is connected to a peripheral compartment, in parallel, containing a network of hollow perforated fibers with defined molecular weight cut-off, and in series to another reservoir that collects the output (excreted) from the central and peripheral reservoir. The inflow of bacterial medium from the central compartment to the peripheral compartment and outflow from the peripheral compartment to the central compartments are controlled by peristaltic pumps. Bacteria are inoculated into the peripheral compartment where they multiply without getting into the central compartment outside of the fibers due to the permeability limitations of the pores. This represents the physiological situation where the peripheral compartment simulates the tissues, the site of infection by bacteria, and the central compartment simulates blood or plasma. The desired drug or test compound is dosed into the central compartment either as a bolus or at a controlled rate. Drug flows into and out of the hollow fibers in the peripheral compartment to the central compartment and is constantly removed from the central compartment into the waste reservoir. The flow rates set in the peristaltic pumps between the inlet and outlet from the central compartment determine the PK profile of the drug following a given dose. The growth and death of bacteria in the peripheral compartment is measured to evaluate the pharmacological effect over time of the drug with the associated PK profile.
The HFIM model has been used to evaluate anti-bacterial effect under simulated human PK of antibiotics, understanding the relationship between dose and dose-schedule in the emergence of resistance to antibiotics, identification of the PK/PD index and its magnitude associated with antibacterial effects like stasis and killing, and prediction of clinical success and failures at approved clinical doses against drug resistant and susceptible strains of pathogenic bacteria. Another important use of HFIM in antibacterial drug discovery is its ability to integrate with Monte Carlo simulations of virtual clinical trials for the prediction of the probability of target attainment (PTA) for clinical doses of antibiotics.
The PK/PD behaviour of antibiotics in HFIM has been shown to be similar in animal models of infection. The HFIM has successfully predicted the microbiological efficacy outcomes in the clinic for many antibiotics at their clinically approved doses.
The major caveats of the HFIM are that it does not simulate the anatomy and physiology of animals for simulation of physiologically based PK (absorption, distribution, metabolism, excretion) in tissues and its high cost.
In summary, the HFIM is a powerful tool that complements animal models of infection and PK/PD modelling and simulation to inform clinical trial design of antibacterial drug candidates to arrive at the optimum dose and schedule for maximizing clinical efficacy and minimizing resistance development.
References:
- Pharmacodynamic Evaluation of Dosing, Bacterial Kill, and Resistance Suppression for Zoliflodacin Against Neisseria gonorrhoeae in a Dynamic Hollow Fiber Infection Model. Jacobsson S, Golparian D, Oxelbark J, Alirol E, Franceschi F, Gustafsson TN, Brown D, Louie A, Drusano G, Unemo M. Front Pharmacol. 2021 May 21;12:682135.
- Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Craig WA. Clin Infect Dis. 1998 Jan;26(1):1-10
- Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it’s not just for mice anymore. Ambrose PG, Bhavnani SM, Rubino CM, Louie A, Gumbo T, Forrest A, Drusano GL. Clin Infect Dis. 2007 Jan 1;44(1):79-86.
- European Medicines Agency (EMA)—Committee for Medicinal Products for Human Use (CHMP). 2016. Guideline on the use of pharmacokinetics and pharmacodynamics in the development of antimicrobial medicinal products (EMA/CHMP/594085/2015).
- U.S. Department of Health and Human Services—Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). 2018. Limited population pathway for antibacterial and antifungal drugs guidance for industry (Draft Guidance).
- Drusano GL. 2017. Pre-clinical in vitro infection models. Current Opinion in Pharmacology, 36:100–106
- Drusano GL, Bonomo RA, Bahniuk N, Bulitta JB, van Scoy B, Defiglio H, Fikes S, Brown D, Drawz SM, Kulawy R, Louie A . 2012. Resistance emergence mechanism and mechanism of resistance suppression by tobramycin for cefepime for Pseudomonas aeruginosa. Antimicrob Agents Chemother, 56:231-242.
- Drusano GL, Neely M, Van Guilder M, Schumitzky A, Brown D, Fikes S, Peloquin C, Louie A. 2014. Analysis of combination drug therapy to develop regimens with shortened duration of treatment for tuberculosis. PLoS ONE, 9:e101311.
- VanScoy BD, McCauley J, Ellis Grosse EJ, Okusanya OO, Bhavnani SM, Forrest A, Ambrose PG. 2015. Exploration of the pharmacokinetic-pharmacodynamic relationships for fosfomycin efficacy using an in vitro infection model. Antimicrob Agents Chemother, 59:7170-7177.
- Lepak AJ, Zhao M, VanScoy B, Taylor DS, Ellis-Grosse E, Ambrose PG, Andes DR. 2017. In vivo pharmacodynamics of ZTI-01 (fosfomycin for injection) in the neutropenic murine thigh infection model against Escherechia coli, Klebsiella pneumoniae, and Pseudomonas aeruginosa. Antimicrob Agents Chemother, 61.
- Tam VH, Louie A, Fritsche TR, Deziel M, Liu W, Brown DL, Deshpande L, Leary R, Jones RN, Drusano GL. 2007. Drug exposure intensity and duration of therapy’s impact on emergence of resistance of Staphylococcus aureus to a quinolone antimicrobial. J Infect Dis, 195:1818-1827.
- Blaser J, Stone BB, Groner MC, Zinner SH. 1987. Comparative study with enoxacin and netilmicin in a pharmacodynamics model to determine the importance of ratio of antibiotic peak concentration to MIC for bactericidal activity and emergence of resistance. Antimicrob Agents Chemother, 31:1054-1060.
- Generating Robust and Informative Nonclinical In Vitro and In Vivo Bacterial Infection Model Efficacy Data To Support Translation to Humans. 2019. Bulitta JB, Hope WW, Eakin AE, Guina T, Tam VH, Louie A, Drusano GL, Hoover JL. Antimicrob Agents Chemother. 63(5):e02307-18.