
The ONCOME could determine the Pharmacodynamics (Efficacy) of Anti-Cancer Drugs
Chemotherapy with targeted drugs in cancer is based on the presence of the abnormally expressed drug target (driver mutations that result in altered receptors or enzymes, or overexpression of the target) leading to dysregulation of biochemical signaling pathways and development of a cancer. The hypothesis is that when this abnormally expressed target is inhibited by the drug it should inhibit or kill the cancer cells, decrease tumour volumes or reduce metastasis eventually leading to increased survival.
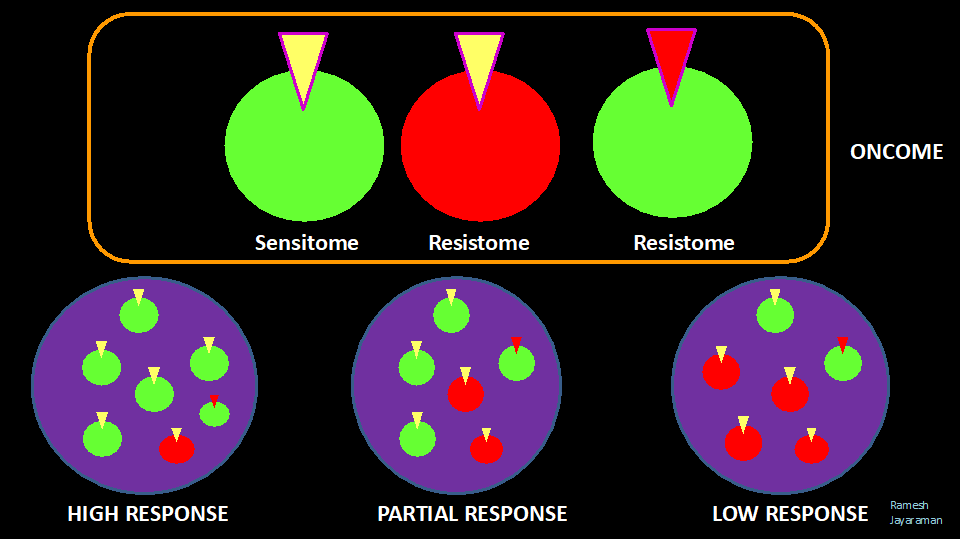
When patients possessing driver mutation(s) are treated with the targeted drug, they show highly variable responses ranging from low to high when monitored for tumor regression and survival. How can this be explained?
A cancer patient can harbor many different types of cancer cells in a primary tumor and metastasized tumors. Since cancer cells grow by acquiring mutations over time and evolution, at any given time, there is a mixed population of cells containing different sets of mutations (clones and sub-clones) in addition to the mutated or overexpressed drug target. Thus even though the primary target is present, there could be other upstream or downstream molecules in the signal transduction pathway that could be either absent, downregulated or over expressed leading to abrogation of inhibition (loss of pharmacological effect). There could also be bypass signaling pathways not connected to the drug target that could lead to escaping the inhibitory effect of the drug.
When a patient is treated with a specific therapy targeting the abnormally expressed receptor or enzyme, some clones are susceptible to treatment (SENSITOMES – sum of all molecular alterations conferring susceptibility to a therapy) and some are resistant (RESISTOMES- sum of molecular alterations conferring resistance to a therapy). The total population of Sensitomes and Resistomes form the ONCOME of the patient.
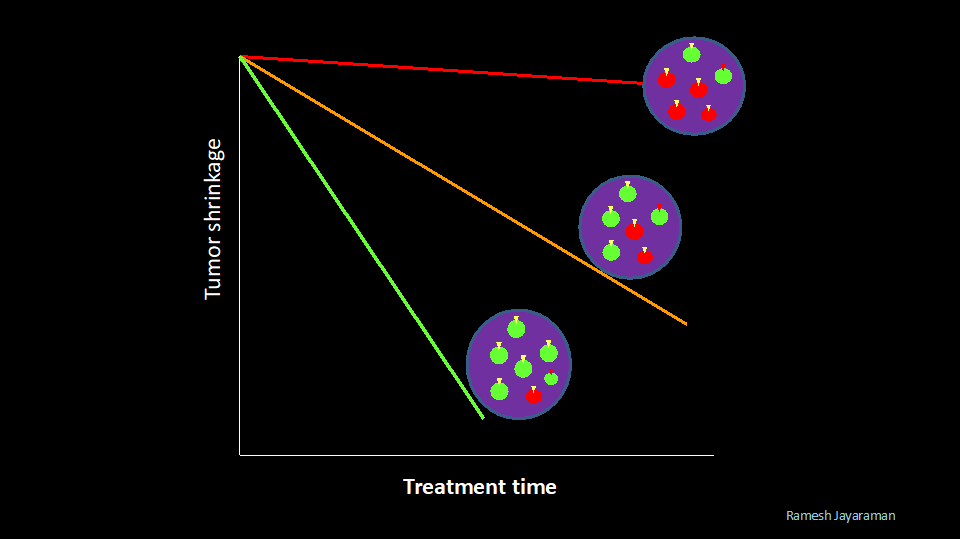
The populations of Sensitomes and Resistomes vary over time (stage) and space (inside tumors – intratumoral heterogeneity and between tumors -Intertumoral heterogeneity). The proportion of Sensitomes and Resistomes in a tumor(s) in a cancer patient could determine the outcome (Objective Response or Clinical Response) of a specific treatment.
The probability of clinical response of a patient to a specific therapy may be predicted using the following equation
Clinical Response = CellSen-CellRes /[CellSen + CellRes]
CellSen = No of Sensitomes in tumor(s)
CellRes = No of Resistomes in tumor(s)
CellSen+CellRes = Oncome of patient
When CellSen>>CellRes the probability of response is high
When CellSen= CellRes the probability of response is stable, constant
When CellSen<<CellRes the probability of response is low
Clinical Response in this context is the probability of a patient responding to a specific targeted therapy as measured by the objective response (tumor growth inhibition or tumor shrinkage) during the course of initial treatment.
The inhibition or killing of cancer cells by a therapy can be described by the following equation
dCellonc/dt = [KgS,i*CellSen,i – Kkill,Sen,i*C*CellSen,i] + [KgR,i*CellRes,i – Kkill,Res,i*C*CellRes,i]
KgS,i = Growth rate constant of Sensitome cell populations
CellSen,i = Sensitome cell populations
Kkill,Sen,I = Killing rate constant of Sensitome populations
KgR,i = Growth rate constant of Resistome cell populations
CellRes,I = Resistome cell populations
Kkill,Res,I = Killing rate constant of Resistome populations
C = Pharmacokinetics of drug
References
- Vogelstein B. & Kinzler KW. 2004. Cancer genes and the pathways they control. Nature Medicine 10 (8): 789-799
- Gerstung M. et al 2020. The evolutionary history of 2,658 cancers. Nature 578(6): 122-128
Ramesh Jayaraman
Director
DoseQuantics Consulting